Nov, 2022
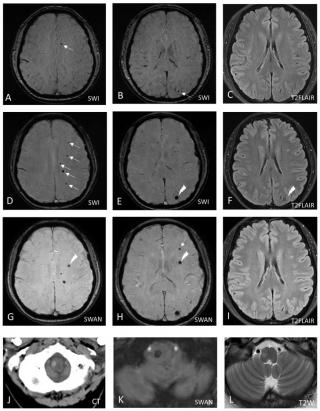
Hereditary cerebral cavernous malformations (CCMs) are characterized by clustered dilated capillary-like vessels in the brain. Autosomal dominant polycystic kidney disease (PKD) is characterized by renal cysts and extra-renal abnormalities. We report a Taiwanese family in which the index case exhibited coexisting phenotypes of both CCMs and PKD. The index case was a 55-year-old woman with known PKD who developed an intracerebral hemorrhage (ICH) in the right medulla. Neuroimaging revealed numerous microbleeds in the bilateral cerebrum and cerebellum. Radiological CCMs were suspected given the absence of other imaging markers of small vessel disease. A comprehensive panel of 183 cerebral vascular malformation genes were investigated through genome sequencing. A novel CCM2 frameshift variant (c.607_608delCT, p.Leu203Valfs∗53) causing a pathogenic premature stop codon, and a known PKD2 nonsense variant (c.2407C > T, p.Arg803∗), were found. Segregation analysis revealed that four siblings were affected by either isolated aforementioned PKD2 or CCM2 variant. Notably, radiological CCMs were exclusively found in siblings who had this CCM2 variant, and bilateral internal carotid artery aneurysms were restricted to one sibling who had the PKD2 variant but not the CCM2 variant. Our study expands the genetic spectrum of CCM2 and demonstrates unambiguous cosegregation of CCM2 and PKD2 variants with their respective phenotypes.